I see that Baran has published a monumental piece of work discussing the conversion of one steroid into another in a linear synthesis of about 20 or so steps and 0.5% overall yield:-
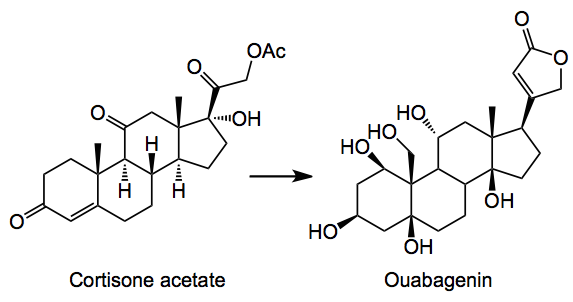
This particular steroid is known for its cardiac stimulation and was first isolated in the late 19th century form the roots of an African tree as its corresponding glycoside the aglycone being isolated by Mannich. From a synthesis point of view they pose some non-trivial challenges for example,the cis-ring junctions a highly oxygenated system and a quaternary center to mention just a few and if that is not enough they form nice complexes with your glassware. Obviously I won’t post the entire synthesis, but will try and pick out some gems (at least I think they are).
One thorny problem is the oxidation of the C19 methyl group and Baran re-visited some of the reported work and came across a synthesis from 1961 by an old colleague of mine which employed a Norrish type II photochemical generation of a cyclobutanol which would then be oxidatively fragmented. Now it has been a long time since I read the words Norrish I and II and it brought back many fond memories of university life This prompted me to go back and refresh my memory about Norrishes! . So the reaction is:-
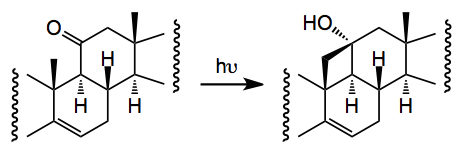
When done in the solid phase the yield is 68% (cf liquid, 43%).
The next gem is shown in the following reaction:-
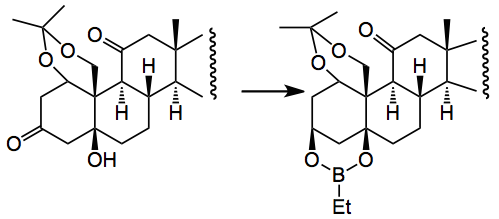
This reduction is completely diastereoselective and you tie up the two OH groups as a boronate ester which acts as a protecting group. This group can withstand some quite exciting reaction conditions, for example, Li/NH3, PPTS, SiO2, O2/Co(acac)2, a fluorination reaction with Selectfluor, another set of oxidation conditions and so is really quite stable. Worth noting this one.
The next thing I noticed was an interesting solvent choice in the following sequence:-
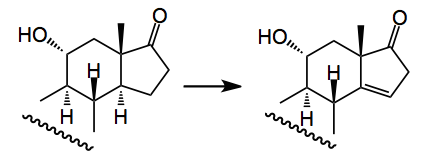
Here oxidation of the ketone with TMSTf/palladium acetate gives the conjugated enone which is “deconjugated” using SiO2/Hünig’s base, this in 55% yield. The solvent for the latter reaction is octafluorotoluene. This apparently minimised the epimerisation of the C14 stereocenter, because the beta epimer cannot undergo isomerisation of the enone. Now where this solvent choice came from is not clear as the referenced literature uses hexane/ethyl acetate mixtures, or toluene. I think this is someones inside knowledge playing a role here. However it is an interesting little sentence in this paper.
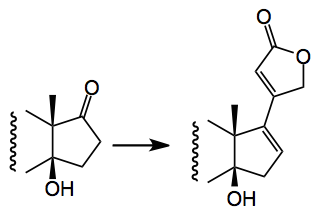
Another difficult problem was the introduction of the butenolide group, in the correct stereochemical fashion. Estrone was used as a model system and after a lot of experimentation they finally got it to work and made the model compound via a hydrazone/boronic acid route:
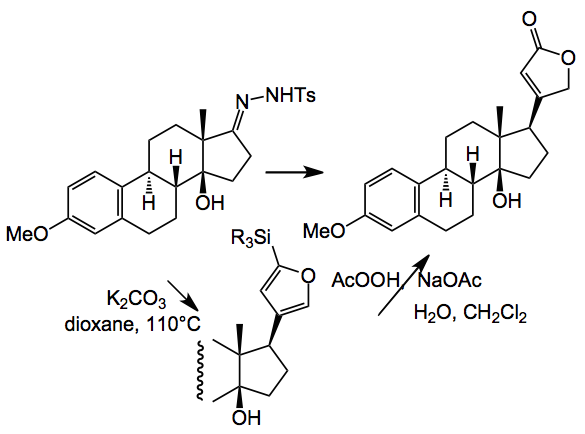
But when applied to the real system this coupling failed! Imagine the feelings of the investigators at this point and you can bet they just did not try it once. They then returned to previously investigated Stille coupling conditions, found during the extensive experimentation, to get this group in place:-
Reagents: 1) 10 x N2H4. 10 x Et3N, CH2Cl2:EtOH, 4:1, 50°C, 5 hr then 3 x I2, 4 x Et3N, THF: 2) 4 x Stannyllactone, 4 x [Ph2PO2][NBu4], 0.15 x Pd(PPh3)4, 3 x CuTC, DMF, rt, 2 hr.
But hydrogenation of the diene always occurred from the wrong face of the molecule. Other methods of reduction were also non-selective. The following sentence from the authors says it all:”We eventually discovered that the use of in situ-generated Co2B led to chemoselective formation of tetrasubstituted olefin”
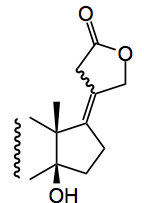
The reductant here is a mixture of sodium borohydride and cobalt chloride hexahydrate in a 2:1 ratio in ethanol. Now if this reductant is Co2B or Co(BH4)2 is not clear to me. The original literature suggests the latter species although complex cobalt hydrides may also be present. It is thought to proceed via hydrocobaltation of the olefin followed by reductive cleavage of the Co-C bond.
This compound was, after extensive screening of organic bases and superbases, converted to the desired product by treatment with Barton’s base:
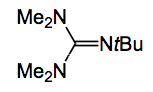
to give a 3:1 mixture of enoates in the desired sense. A quick blast with HCl removed all the protection providing ouabagenin in 0.5% overall yield.
This post only highlights the things I spotted there are probably many others gems tucked away here. This is a wonderful example of organic chemistry. Lots of “simple reactions” that turn out to be not so simple in the real world. I applaud the perseverance of the group in surmounting the problem(s), especially at the end of the synthesis. I recommend this paper as a wonderful piece of work, a good read. Remember to get the supplementary info. lots of detail in there as well.
![]()