Previously I mentioned the importance of isolating and identifying side products. The synthesis of the 3D compound produced lots of them and we chased them all down. Particularly interesting were those isolated in the mother liquors of the drug substance re-crystallisation. To stress the point there were quite a few of them but not really in large amounts, a tribute to the process that it didn’t generate larger quantities!
The last step in the synthesis was the protecting group cleavage, as it is in many cases. This turned out to be somewhat trickier than I expected and is quite complex especially when trying to maximise product and minimise side product formation. So I was dealing with the cleavage of 3 silyl ethers. The literature is unclear on the best method for this transformation. Some authors use HF/pyridine, some HCl/MeOH, others employ HCl/THF, or para-toluenesulphonic acid. There is no obvious reason to be gleaned for utilising differing acidic systems, especially within the same publication! The actual reaction is below:
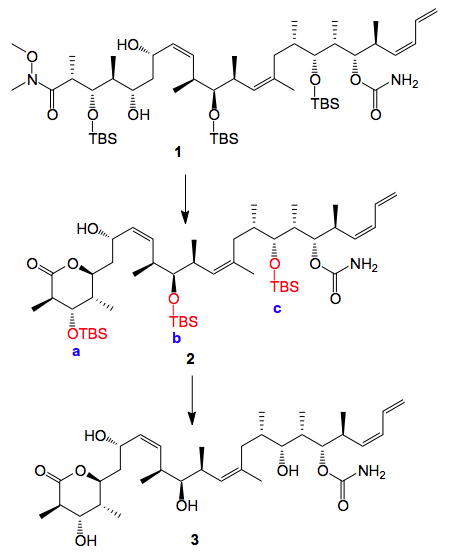
Methanol/HCl was used and the first thing that happens is the cyclisation of 1 to give 2. This compound oils out of the reaction mixture and distributes itself around the walls of the reactor. During the reaction monitoring this is not obvious and only becomes so after work-up. This problem may be avoided by continually washing the walls of the vessel with methanol in order to maintain 2 in solution and adding the HCl in portions over several hours, if this is done 3 can be isolated in 70% yield after reverse-phase chromatography. The slowest silyl group to cleave is that at the C3-position, silyl group a in the scheme, and we were able to isolate this compound 4 after column chromatography of the reaction mixture.
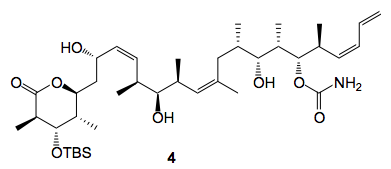
Forcing the reaction conditions results in significant formation of side products, the isolation and formation of these will be discussed below.
If one takes isolated 2 and is a bit more brutal with the acidic cleavage several side products can be isolated by chromatography of the mother liquors of the crystallisation of 1.
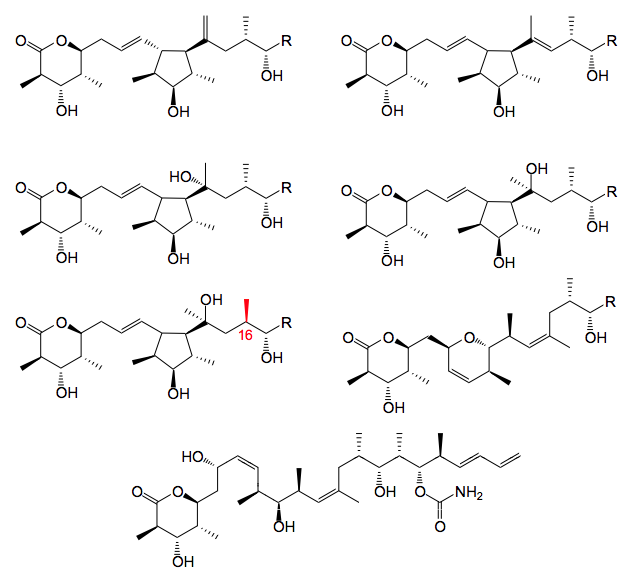
A couple of comments here: The structures were all determined by 1-H-NMR and mass spec. analysis. Their formation can be rationalised by realising that the nucleophillicity of the C13-C14 double bond plays a significant role certainly due to it’s proximity to the potential cationic site. The allylic cation formed can either be trapped by the double bond leading to the cyclopentane rings or the C11 – hydroxy group producing dihydropyran. Marked in red the C16 methyl group apparently has the opposite configuration to that observed in discodermolide. This stereocenter is formed with an aldol reaction from earlier in the synthesis. It is reasonable to assume that traces of the diastereoisomer have been formed and carried through the synthesis, although we did not observe the corresponding C16 (R) isomer of discodermolide.
One more side product was found, the trans diene. This compound runs immediately after discodermolide on HPLC and unfortunately co-crystallises with it. It is apparently formed in small amounts during the acid cleavage of the silyl groups although its formation as a consequence of the Nozaki-Hiyama-Kishi/Peterson olefination reaction cannot be ruled out.
So it is always worth it, to chase after your “side products”. Do this and you may be pleasantly surprised.
![]()