Ketones are always useful building blocks in organic synthesis, not just because of their utility in aldol reactions and other enolate chemistry but because the reactivity can be tuned by Umpolung, a concept that has been around for many years. Typically a dithiane can be sequentially alkylated and subsequently cleaved to form unsymmetrical ketones, as so elegantly described by Smith in several of his natural product synthesis.
This week see the deployment of a new system, CLAMP, by Sarpong from the University of California. This is the equivalent of a carbonyl dication equivalent:
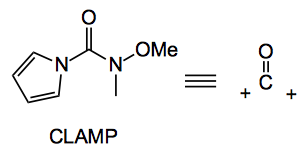
The idea is described by the authors “In order to effect the selective formation of unsymmetrical ketones, an electrophilic carbonyl linchpin must feature two nucleofuges, each of which generate differentially stable tetrahedral intermediates upon sequential monoaddition of organometallic nucleophiles. Specifically, initial reaction of a carbonyl dication synthon with the first nucleophile equivalent should afford a tetrahedral intermedi-ate that persists throughout the addition but whose rate of collapse can be modulated by experimental conditions. Controlled collapse of the first tetrahedral intermediate generates a new carbonyl electrophile (acylium ion synthon) that is available for reaction with a second organometallic nucleophile. Ideally, persistence of the tetrahedral intermediate derived from the second addition prevents overalkylation at the electrophilic carbon, preserving the ketone oxidation state.“
So how do you make this wonder reagent? Well you won’t find it in the paper, anywhere, not even a reference to it! This must be one of these “Mary Poppins” reagents, click your fingers, say the magic words and the stuff arrives out of thin air and does it’s thing.
Sorry but this is a lot of fermented bovine manure. Why did the referees not insist it was included in the main paper? You will find it, in the supplementary information, I reproduce it here, without permission:
N-Methoxy-N-methyl-1H-pyrrole-1-carboxamide (2, CLAmP)
A solution of N-methoxy-N-methyl-1H-imidazole-1-carboxamide (2.25 g, 14.5 mmol) in anhydrous acetonitrile (40 mL) was stirred while pyrrole (0.97 g, 15 mmol) was added in one portion. The mixture was stirred at room temperature while DBU (2.2 g, 15 mmol) was added rapidly dropwise and stirring was continued for 3 hours at room temperature. The reaction mixture was concentrated in vacuo using a rotary evaporator and the resulting brown oil was partitioned between EtOAc and sat. NH4Cl (aq). The organic layer was collected and the aqueous layer was extracted with EtOAc (2x). The pooled organic layers were washed with brine, dried over MgSO4, and concentrated in vacuo. The crude product was purified by flash chromatography (10→20% EtOAc in hexanes) to afford the title compound (1.82 g, 81%) as a colorless oil. The title compound becomes brown upon prolonged storage under ambient conditions, but can be repurified by chromatography.
1H NMR (500 MHz, CDCl3) δ 7.41 (t, J = 2.4 Hz, 2H), 6.26 (t, J = 2.4 Hz, 2H), 3.68 (s, 3H), 3.37 (s, 3H).
13C NMR (151 MHz, CDCl3) δ 152.1, 121.3, 111.2, 60.8, 35.2
FT-IR (NaCl, thin film) νmax 2973, 2937, 1685, 1421, 1382, 1259, 1109
HRMS-EI (m/z): [M]+ calcd for C7H10N2O2+, 154.0742; found, 154.0744.
For the synthesis of this starting material, see S. T. Heller, R. Sarpong, Org. Lett. 2010, 12, 4572-4575.
Note it is not stable on storage, it goes brown. Chromatography as well!
I’m going to stop here as this ruined what could have been and should have been a useful methodology paper.
![]()