This week’s offering deals with a reaction which I experimented with several years ago (more later), the well studied [3+2] cycloaddition of azomethine ylides with alkenes producing pyrrolidines. Normally the ylide is derived from the imine of an amino acid however Waters and his group now report a novel entry to produce azomethine ylides starting from allylic amines and glyoxals.
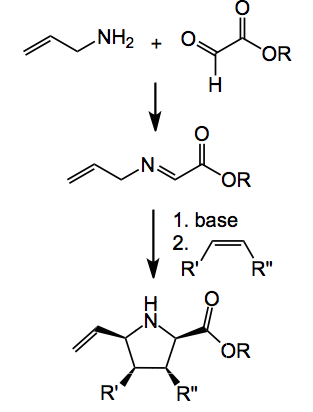
Normally these imines are not acidic enough to form the ylide, however, in the presence of a metallic catalyst triethylamine is sufficient to form the ylide. Silver acetate is the catalyst of choice here and Waters used 10 mol%. By carefully optimising the reaction conditions he was able to obtain the pyrrolidine after cycloaddition with the ylide derived from benzylamine and phenyl maleimide as a single diastereoisomer in good (86%) yield. One example of the kinds of pyrrolidine he was able to produce is given below.
So quite a useful method and it removes the reliance on amino acids for the success of this chemistry.
My interest in this chemistry was in obtaining the following compound, for reasons I won’t go into here:
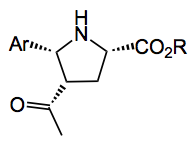
But I was stuck with the amino acid ester approach, but was able to prepare a large series of imines from ArCHO, most of which were stable and if you chose the correct ester, i.e. t-butyl the imines were crystalline. So using 1.0 – 1.5 equivalents of AgOAc I obtained a 40 – 50% yield of the pyrrolidine after reaction of the imine with methyl vinyl ketone. As was also noticed by Waters a side product is formed namely the N-alkylated compound formed by conjugate addition of the product with methyl vinyl ketone. In my case I was able to crystallise product and the N-alkyl compound remained in the mother liquors.
Silver acetate is expensive and I used up to 1.5 mol equivalents! However, Zhang reported a catalytic version of this reaction, not with methyl vinyl ketone. Using his conditions I was able to reduce the silver acetate to 5 mol% simply by adding 5 mol% of triphenylphosphine which solublises the silver salt. The pyrrolidine came out in 50-60% yield. One minor problem, I got a wonderful silver mirrored flask as a side effect. I imagined this on a plant scale in a 1000L reactor it would not have made many friends.
Of course the compound(s) I made were achiral and only one diastereoisomer was formed due to the cis addition. Chirality is always desirable so I looked at (R) and (S) – QUINAP as the phosphine source (5 mol%).
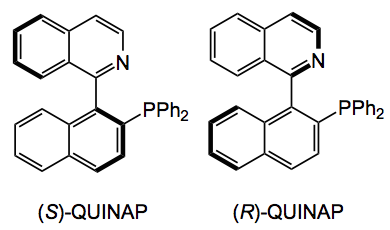
So this produced the pyrrolidine, after reaction with methyl vinyl ketone and the t-butyl aryl glycine imine in 40 -50% yield with an ee of 60-70% after reaction at -20°C, the result being the same with either the (S) or (R) the QUINAP. One re-crystallisation brought the ee up to 93% but further crystallisation did not improve this it just caused losses of material.
Now QUINAP is probably more expensive than silver acetate as its synthesis is very difficult and the resolution employs some horribly complex chiral palladium salt (whose structure I won’t attempt to draw here) and fractional crystallisation. So I examined many catalyst/ligand systems; the Trost ligand, Josiphos, quinine, Cu/ferrocene, Pflaz’s ligands, Carierra’s QUINAP variant, zinc triflates and so on. No luck, no reaction or reaction and no ee! So I was stuck with QUINAP. I forgot the chiral system and resorted to preparative chromatography using a Chiralpack column and supercritical CO2 as a mobile phase with some solvent system whose composition I forget. Anyway out came both enantiomers and eventually I had both of them in >10g quantities with ee’s of >98.8%.
So really this synthetic method is really great and works well. Since I was interested several newer publications (one from a South American group, this year in JOC) have reported really high %ee’s and I never got round to checking them out in my system, mainly because the project died. Typical for industry, you get a reasonable result and the project dies because of something going on elsewhere in the development chain.
Anyway a useful ASAP from the Waters’ group it will be nice to see how it develops in the coming publications.
![]()
Hi there; a colleague passed on your blog because of the Quinap connection; nice that you got the Chemistry to work eventually.
But your comments are off beam – “synthesis is very difficult” fresh graduate students used to make 15 g of each hand for Strem until they started charging ridiculous profit-driven prices that only Pharma could afford – “horribly complex chiral palladium salt” a standard in ligand evolution and works exquisitely in this case – “fractional crystallisation” -not in any manuscript, where did that come from?
John B
Thanks for the comment.
The QUINAP synthesis is by no means trivial. According to your Tetrahedron Asymmetry paper from 1993, 4, 743 you have a 10 step synthesis with a yield to the diastereoisomeric complexes of 13.5%. Unfortunately you do not quote the yields of the crystallisation in that paper. But I will assume 100% that is 50% (R), 50% (S) thus that 13.5% becomes 6.8% for a optically pure diastereomer, followed by a “decomplexation” with 88% yield, i.e. the overall yield of the pure (R) or (S) QUINAP is 6%!
You are performing a resolution by crystallisation of diastereoisomeric salts or in this case palladium complexes by exploiting their differing solubilities in a given solvent, by definition this is fractional crystallisation. In this case you were lucky that the solubility differences were large in the solvent allowing a complete resolution with 1 crystallisation, although you do not quote a yield for this step.
Now imagine you are going to do a 100 Mole reaction, at 5 mole% (which I used) you need 5 mole of QUINAP (2.2 kg) at 6% overall with a 10 step synthesis to the pure enantiomer requires 36 kg starting materials. As you use a 2:1 ration of racemic QUINAP to the chiral Pd complex means 72 kg of starting materials. Ever run a Grignard at approaching the 100 kg scale? Not trivial. Or a Suzuki at this scale? Not trivial. For example, you said in the publication that the first coupling reaction, when run at the 1 g scale gives 96% yield, in the experimental part that same reaction when run at a 60g scale gives 78% yield, why is this? This is not a trivial problem to solve.
This is not simple chemistry with Pd catalysed coupling reactions not to mention the Grignard reaction, the BBR3 cleavage (which often produces ring bromination side products) and a silane reduction. I won’t even start on the use of trimethylborate and chloroform, dichloromethane etc. In a plant environment this whole QUINAP sequence would be very difficult to do given the constraints of the production environment.It can be done but would require significant investment in time to evaluate the process correctly.
I have not searched the chiral Pd complex that is used for the resolution so I have no information as to its synthesis or availability. Further there is no mention of its recovery and re-use in your paper, can this be done?
I believe you have also patented this synthesis and the compounds involved, this was a wise move
I was never able to recover the QUINAP, I don’t know if this can be achieved.
So I don’t accept that my comments are “off beam”.
As an aside: I believe you have also patented this synthesis and the compounds involved, this was a wise move and only increases the non-triviality of the synthesis as industry, being somewhat miserly, would certainly seek alternatives were it to be employed in any commercial application.
Here is a paper from 2003 published in Organic Process Research and Development.
http://pubs.acs.org/doi/pdf/10.1021/op034007n
The synthesis and resolution of Quinap is described on a large scale.
Thank you for the link. I did have this paper in my collection. This is certainly a larger scale than the original publication. The synthesis of (RS)-QUINAP is carried out on a 0.16 mol scale based on the triflate. I note that the reagents were added in one portion to the catalyst! This step is really poorly defined, in my opinion. For example, “….diphenylphosphine (9 g, 48 mmol) was added to a solution of NiCl2(dppe) (8.51 g, 16.1 mmol) in DMF (200 mL) at room temperature, and the resulting dark brown solution was heated for 0.5 h”. no temperature indication given here. Further “transferred in one portion to the reaction flask via cannula” this is rather dangerous as no indication is provided as to exothermic processes which may be operating here, although the mixture is latterly heated to 100°C. This on a much larger scale would require extensive calorimetric experimentation to determine the process safety. With 68% yield one should ask what is the rest of the mass?
Also in this publication there is the use of ether in one of the steps and dichloromethane in most of the steps. The Grignard reaction is also poorly described with no temperature reported during the drop-wise addition of the bromide, did it reflux? Did it require cooling, I assume it is exothermic.The yield is 78%, what is the rest of the material? There are other points that I could raise.
While this is done on a larger scale than the Tet. asymmetry publication, with slightly better experimental details I don’t really consider it a real significant improvement.
The synthesis of this compound is not trivial.
I agree with you there are a number of steps which are not trivial and also the amount of steps is major drawback using this ligand on industrial scale. That is even before IP is considered. Solvents like Diethyl Ether and DCM while dangerous at scale, are still cheaper and more efficient than purification by column chromatography.
For full disclosure I am a PhD student at University College Dublin, Ireland working on the area of P,N ligands and have recently contributed to a perspective of Quinap and similar ligands with John Brown.
http://pubs.acs.org/doi/abs/10.1021/jo500512s
The resolution step is a key component in the Quinap synthesis and resolution by Palladium salts is the main method used in these types of P,N ligands unless using Carreria’s Pinap which is surprisingly similarly priced to Quinap. A new methos has been developed by Stoltz which may prove amenable to large scale
http://pubs.acs.org/doi/abs/10.1021/ja409383f
The method described in the Organic process research and development paper uses half the amount of Pd as the earlier tetrahedron paper (when comparing moles used).
Also it has to be stressed that this synthesis in the Organic process research and development is conducted by academic scientists, not process chemists. An exothermic study would be key from a safety perspective and also the concentration for each step would be optimized to save solvent. However, few catalysts are used industrially unless they are ferrocene based (Josiphos, Taniaphos etc). Trost ligands and Quinine.
Thanks again for the comment. I shall obtain a copy of the perspective. The use of diethyl ether on scale requires very special facilities, which were present where I worked. No they were installed several years (decades) ago and infrequently used. Thus I don’t think they ever paid their way, so in that respect CC may actually be cheaper:)
I realise that the resolution employed less Pd that originally and this may actually be the easiest step in the entire sequence.
I had absolutely no success with PINAP, I could not reproduce the literature and gave up.
While I appreciate that these are “academic people” but if they publish something like this in such a journal some mention/comment of the thermal aspects of the reactions deserves to be made. Just saying heating for 30 minutes without even mentioning the temperature it was heated to shows a lack of attention to what may be an important detail. This part of the synthesis, the in situ preparation of the active catalytic species, is of major importance for the success of the reaction and deserves more detail.
As I said the lowish yields of some of the steps contribute to my criticism of this synthesis. Once again the mass balance of the reaction should be accounted for. That is not process chemistry but good technique.