This week we have a publication with significantly fewer than 5154 authors, only 6, from Gademann, from Basel and Eberl who resides in Zürich. They have put aside the old rivalry between the two cities and describe their work on the isolation, structure elucidation and total synthesis of Kirkamide.
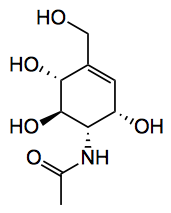
This compound is “from the bacterial leaf symbiont of the Rubiaceous shrub Psychotria kirkii (P. kirkii)” and is, amongst other things, an extremely effective insecticide. The isolation used a NMR technique to detect the presence of the new compound in the fractionation of the plant extract, quoted: “Since aminocyclitols display a characteristic methylene proton signal at around 6 ppm in the 1H NMR spectrum, we opted for an unusual genome-based, NMR-guided fractionation approach. In order to overcome sample limitation, we used a 1.7 mm micro-cryoprobe 600 MHz NMR spectrometer in the early stage of the discovery process. Submicrogram amounts of sample were sufficient to record a 1H NMR spectrum that confirmed the presence of a C7N aminocyclitol in the extract”. Fractionated was 160 mg using a semi-prep. RP-HPLC. Final purification of the new compound was achieved by combining HPLC with Cu(II) coated prep-TLC plates, which is a technique reported for saccharide separation.
Turning to the synthesis. Starting point was the N-acetyl-D-glucosamine which was converted to the enol in 5 steps:
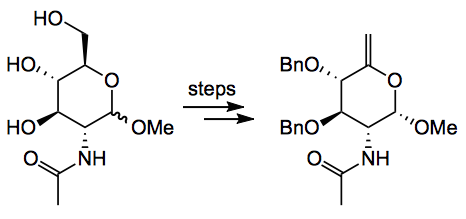
Yield was 18%. This sequence required a cumbersome protecting group swap necessitated by the nature of the first intermediate (an iodomethyl derivative) not allowing direct benzylation! Also note the anomeric stereochemistry. The key reaction, a mercury catalysed Ferrier carbocyclisation,
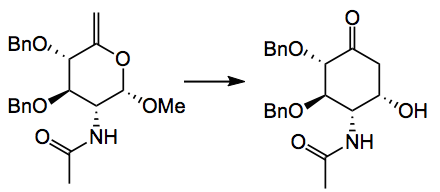
“HgSO4 (0.452 g, 1.52 mmol) was added to a solution of the benzyl protected olefin 5 (2.24 g, 5.64 mmol) in 1,4-dioxane (56 mL) and aq. H2SO4 soln. (5.5 mM, 28 mL) that have been degassed 10 min with argon. The mixture was stirred at 60°C in the microwave for 15 min, diluted with CH2Cl2 (600 mL), washed with brine (2 x 400 mL), dried over Na2SO4 and evaporated under reduced pressure. The solids were washed with Et2O to afford 6 (1.74 g) as a white powder”.
This corresponds to about 80% yield, this white powder was silylated to give the corresponding OTBS anomer (63% over 2 steps). Assuming the silylation goes in >95% yield the 1.74g is not as pure as one might imagine! Or the silylation is not very efficient. No details as to purity were provided. No details about the microwave were provided. No details about the heating or the internal temperature were provided. So very little useful information was provided. A final sequence of 4 steps delivered the natural product in about 25% yield. These steps contained a microwave assisted Stille coupling and a sodium liquid ammonia benzyl group cleavage. A further comment from the authors “All of the synthetic steps up to the final deprotection were amenable to gram-scale preparation, which demonstrates the robustness of this route.“
I’m sorry to say that I find this last comment a bit annoying. A gram scale synthesis of anything does not demonstrate robustness. And with the sparsity of detail it is anything but robust. What is wrong with the last step? The penultimate step (desilylation) was done using 1.38g, for the last step they used 40 mg.
This is not a total synthesis publication, the chemistry is only a means to an end, which is OK. However, I get the impression that the compound was assembled ASAP. With a bit more thought and a few more details the chemistry could also have been a highlight of this paper instead of playing second fiddle.
The synthesised material demonstrated the required biological activity.
![]()