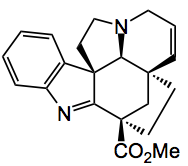
This weeks highlight for me was a publication by K. Lee and D. Boger of Scripps in California dealing with the total synthesis of two alkaloids, (-)-Kopsifoline and (-)-Deoxoapodine. This synthesis once again demonstrates the power of cycloaddition reactions to generate multiple rings with stereochemical control.
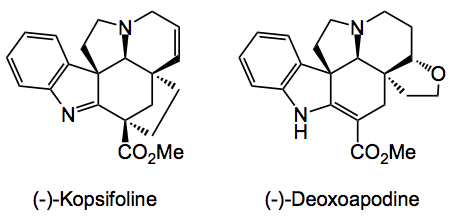
Just look at the complexity here, as my Phd supervisor said “bristling with functionality”. This is a typical Boger problem, over the years he has synthesised many alkaloids notably his work in the vinblastine area sticks in my mind, the groups home page has a complete list of the publications, some 550 over the years.
Returning to the current synthesis: Dale recognised that by astute selection of a common precursor he could access several (structurally similar) alkaloids by simply varying the chemistry around the molecule after the key cycloadditions had been successfully achieved. The basis of the approach and key to the synthesis of the underlying Aspidosperma skeleton of these alkaloids is a powerful intramolecular [4 + 2]/[3 + 2] cycloaddition cascade of a 1,3,4-oxadiazole that provides the fully functionalised pentacyclic ring system in a single step.
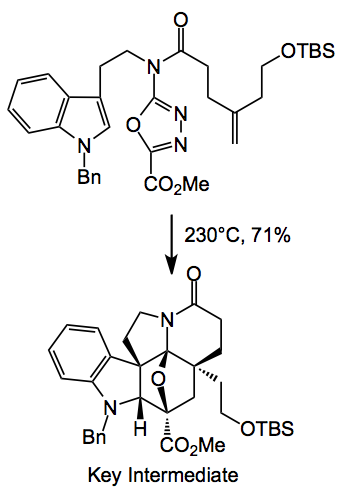
So there it is as simple as that. Just four steps from tryptamine and you get a degree of complexity impossible to obtain in any other way. The key intermediate is a single diastereoisomer. Reductive cleavage of the oxygen bridge with sodium cyanoborohydride delivers the corresponding α-hydroxy ester which is then converted into a methyl dithiocarbonate ready for radical desulphurisation with tri-n-butyltin hydride (Barton-McCombie deoxygenation). Note the change in stereochemistry of the ester. This provides the basic ring system of (-)-kopsifoline. After simple functional group manipulations and a resolution you’re almost done:
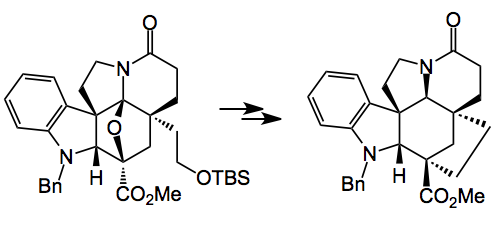
Change the OTBS into an iodide, cyclise, convert the amide to the thioamide and Raney nickel treatment gives a tetrahydro (-)-kopsifoline (above) a compound not yet observed from natural sources.
Back to the key intermediate. After reductive opening of the oxygen bridge a protecting group swap from N-benzyl to the carbamate and a resolution followed by xanthate formation, Chugaev elimination and provides compound 1:
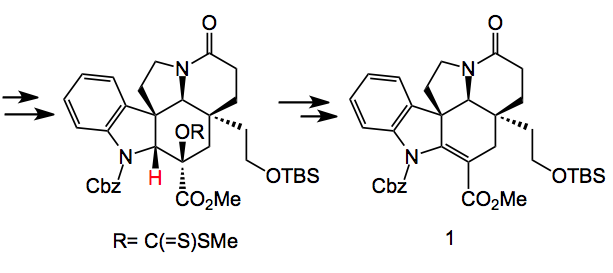 Note the N-protection swap. The carbamate activates the β-hydrogen (shown in red) and facilitates the syn elimination providing the more substituted and more stable olefin. Further manipulation of compound 1, for example, α-selenylation, oxidation to the selenoxide and elimination places a C=C in conjugation with the carbonyl group. Resolution and 6 further steps provides (-)-kopsifoline. This compound was isolated as a hydrate, an interesting fact as it has not yet been isolated from natural sources.
Note the N-protection swap. The carbamate activates the β-hydrogen (shown in red) and facilitates the syn elimination providing the more substituted and more stable olefin. Further manipulation of compound 1, for example, α-selenylation, oxidation to the selenoxide and elimination places a C=C in conjugation with the carbonyl group. Resolution and 6 further steps provides (-)-kopsifoline. This compound was isolated as a hydrate, an interesting fact as it has not yet been isolated from natural sources.
The synthesis of (-)-deoxoapodine proceeds via one of the intermediates from the sequence described above:
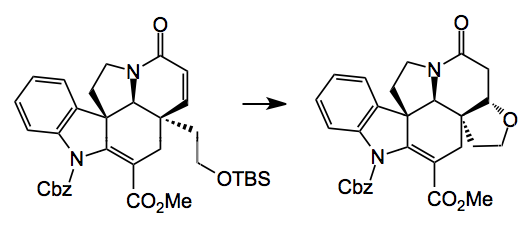
By simply removing the TBS group the conjugate addition product is isolated as a single diastereoisomer in 70% yield. Reduction of the amide carbonyl with borane.THF and a Lewis acid removal of the Cbz group (BF3.OEt) gave around 40% yield of (-)-deoxoapodine.
Once again a short description does not really do justice to the effort applied in this synthesis. Just preparing this post was hard enough, the chemistry is even more challenging. Lots of good reagent and functional group chemistry to be learnt here. But, at least these compounds are really new natural products.
![]()