After spending some years in medicinal chemistry (CNS) I moved to chemical development. Now this change I can recommend to everyone. In med. chem. you are just another tool for the biologists churning out compounds (methyl, ethyl, futile). I think this says it all, one of my biological colleagues asked me for more of a particular compound and added “make it more soluble the next time.”
You don’t get a real chance to use your chemical knowledge and training, you know all that stuff you learnt at university, some of which may have gone into a thesis. Now, in chemical development that is completely different. There you can actually apply your knowledge to scores of problems, even using physical organic chemistry! For me it was like a breath of fresh air and it re-vitalised my organic chemistry tick. At that time we were responsible for everything, reservation of the pilot plant equipment, ordering the starting materials, carrying out use tests of material from new suppliers1, organisation of all the analytics, all the lab work to produce a lab procedure, all the pilot plant supervision including writing a pilot plant procedure, all the safety studies, proposing and carrying out alternative routes, when required, in fact, everything one can think of we were responsible for. It all boiled down to quality, quantity and delivery (on time).
In a previous post here I recalled some of my experiences my first project, now I recount the second. This compound could loosely be called a dinosaur, it had been in development for more than a decade or so and the chemistry, as we will see, had some bite to it. It was a 9-step synthesis of which I had the first 4 steps. The steps under my supervision were; 1) an acid catalysed esterification of a benzoic acid to its methyl ester, 2) reduction with sodium borohydride, 3) conversion of the benzyl alcohol to the benzyl chloride, 4) conversion of the benzyl chloride to the benzyl azide.
A good friend and colleague, not to mention an excellent chemist, had largely completed the chemical development; there were just a few more things to look at in the lab. My role was to produce quantities in the pilot plant for clinical supply and formulation validation. This, of course, meant lots of material had to be produced. We had to obtain around 7000 kg’s of drug substance by a date given by the start of the clinical trials. Calculated was 1 month for release of the drug product by QA, which meant for me I had to start in January and deliver my last step by the end of September. Here is the plan I generated at that time.
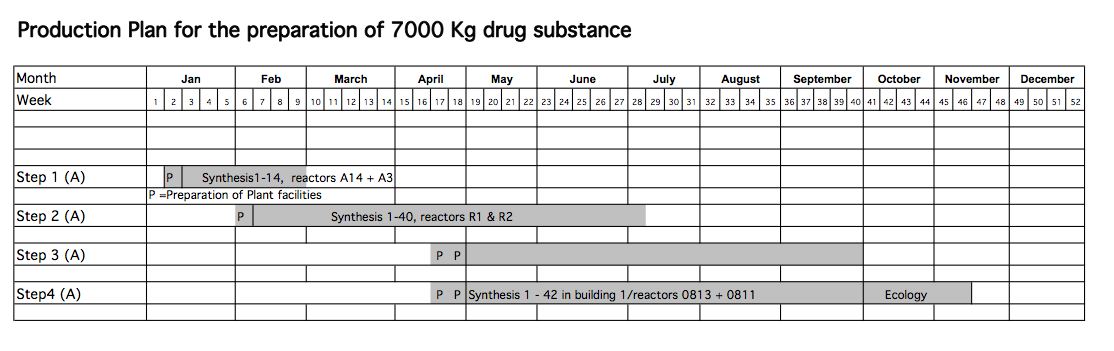
The esterification was easy: it was done in a 2-phase system, hexane/methanol and catalysed by conc. sulphuric acid (extractive esterification). We did 16 batches of 528 kg of the benzoic acid using 238 kg 98% conc. sulphuric acid per batch. After a bicarbonate neutralisation, phase separation and distillation of solvent I got 614 kg of the ester. I did it 16 times, the average yield was 95.75%, and 9145 kg was prepared and used in the next step. It had some residual hexane in it,, about 10% per batch, but this was not a problem for the reduction. The hexane distillate was re-used each batch, just being replenished with fresh material to make up the losses.
Dimethyl ether formation was not a problem. It maximised at a production rate of 500 mg/minute after 1 hour then dropped away to <50 mg/minute after the regular reaction time was over. Not bad for a 528 kg batch and certainly not a hazard.
In contrast to med. chem. at least you can see what you get from your reaction, 16×1 cubic meter containers were used to store the stuff until we started the next step. Remember the plan, it was overlapping so we started step 2 about halfway through the step 1 campaign so things were getting more complicated.
The borohydride reduction was a bit trickier. I did 42 batches of 200kg of the ester as a 90% solution in hexane corresponding to around 1 kMol of starting material plus 8 more batches as “use tests”. Everything was tossed into a 1000 L reactor, warmed to 55-57°C and methanol added via a membrane pump over an 8 hour period.
There is still not a good system for the introduction of such solids as sodium borohydride to a reactor. Borohydride is very hydroscopic and it blocks up the machine. We tried pellets, and these bags that dissolve and release the reagent, but then you just get mushy plastic bag in your product. Neither was as good as the powdered stuff. So in the end we just quickly shovelled it in through the manhole. The hydrogen liberated was vented with a nitrogen flow via a cold (-30°C condenser to trap THF) to the roof via a specially designed pipe with a non-return valve built in so no flames could return and cause a nasty experience.
We set up the first reaction and got it going, then the operator turned to me and said “I’m off for my coffee break now, if anything happens press the red button.” And with that left me standing there praying nothing would happen. Of course he was joking, I hoped, sure enough another operator arrived laughing at my obvious relief. Just as well he came. I looked in the reactor to see a wall of foam coming up to meet me, alarms started to go off as we had about 0.5 Bar overpressure in the reactor which was rapidly increasing. The hydrogen-venting pipe was blocked. So everything was shut down (except the stirrer) and fortunately the foam was contained. Now, it turned out that this pipe had not been used for some months. We went up on the roof and blew a terrific pressure of nitrogen through it from below, and saw several dead birds and various other bits of detritus shooting out of it. Thank God for the non-return valve. The blockage problem solved they placed a steel net over the top to prevent this happening again although why this had not been done before I don’t know. Anyway I went off to change my underclothes and when I got back we started the thing up again and it seemed to be ok this time.
The safety department recommended that, if the reaction got out of control, we should dilute the reaction rapidly mixture by pumping in THF, so a 500 L container under 1 Bar pressure was connected at all times during the reaction, fortunately we never had to use it. This was a safety measure because during DSC experiments we observed a long flat exothermic decomposition between 75°C – 500°C. Our requirements said the reaction must be carried out 20°C below the start of the decomposition temperature which is why we did it at 55-56°C. In any case it would not have been critical, as the maximum temperature of the synthetic reaction (132°C) would not have been reached even if all the THF had evaporated in the case of a condenser problem, the evaporation of the THF would cool the system down. The total energy under that curve from 75° – 500°C was around 700 kJ/kg. Note here the time to maximum rate of the decomposition reaction was estimated at >24 hours, which was fine and meant we would just about survive and the blast proof windows would live another day.
The work-up was interesting; it involved a solvent change to toluene followed by aqueous extraction. So off we went and distilled off the THF/MeOH down to a predetermined volume in the reactor. Note if you applied the vacuum too hard you obtained another foamy mixture advancing its way up and out of the reactor. This was fascinating to watch but I was used to foam by now I knew it would stop which it did, just in time before it reached the condensers. Toluene went in, toluene/THF/MeOH came out and more toluene went in and toluene/THF/MeOH came out, more toluene went in this time followed by water. Now if you did not stir for long enough (2 hours) the aqueous layer was on the top! But I didn’t fall for this one, after the required time the phases swapped over and I obtained a normal water/toluene/product mixture. Off came the water, which was re-extracted and the combined toluene layers evaporated to dryness. All the toluene from the extractions was re-cycled, and used in the next operation for the extraction checking to make sure there was no accumulation of any side products (which, of course, there was not). Filtration of the distillation residue to remove floating white solids provided the product.
So, after nearly 5 months we ended up with the benzyl alcohol. The mean yield was 96.88% corrected for the concentration of the product (as about 3% toluene remained) and we produced 7098 kg. With this we were ready for the two most critical steps in the sequence, the formation of the benzyl chloride and finally the benzyl azide. That will be the topic of the next missive in this small series.
Notes:
1 A use test is employed in order to investigate material from potential starting material suppliers: The compound must meet the given specifications and perform in the series of following reactions to deliver the desired product according to its specifications.
![]()