Sorenson and his group have described an enantiospecific synthesis of the sesquiterpene Jiadifenolide which is full of interesting chemistry.
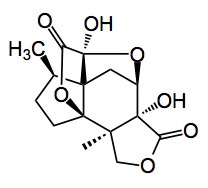
Jiadifenolide
Quite a complex structure containing two γ-lactones, a 5-ring hemiketal 7 contiguous stereocenters all tied up in a novel cage-like structure. So where does one start with such a molecule? A good place is to employ the pattern recognition described by Danishefsky. This method “relies upon the identification of moieties within the target, which are associated with reliable chemistry, and can serve to facilitate progress to the target“. So Danishefsky, for example, recognised the presence of elements stemming from α-phellandrene in the elutherobin system and proceeded to synthesise the latter from the former. So this together with the usual reterosynthetic analysis provides a good basis for the synthetic direction.
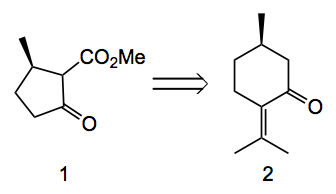
Application to the jiadifenolide case provides the following β-ketoester 1, providing the 7 carbon atoms of the far left cyclopentane ring, which can be related back to (R)-(+)-pulegone 2:
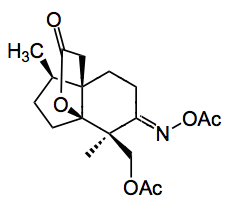
This analysis leads ultimately to the core problem of this synthesis, namely a C-H oxidation of the methyl group (in red) of the advanced intermediate, compound 3.

The DG in this structure is a directing group yet to be defined. Ideally this oxidation needs to incorporate an oxygen atom or a functionalised O atom for example acetoxy.
The synthesis of 3 started with a Robinson annelation of 1 to produce the following enone 4, in 82% yield:

This was further manipulated in 6 straightforward steps to compound 3 in which the DG is now a carbonyl group. The oxidation procedure for functionalisation of the methyl group of 3 was now examined. They chose a method described by Sanford which involves acetoxylation of unactivated methyl groups directed by a neighbouring oxime catalysed by palladium acetate. This afforded the oxime acetate as a 1:1 mixture of both possible methyl oxidation products, which were easily separated by chromatography to give the desired compound in 22% yield.
Other methods were examined however the result was not improved and in the event was actually worse. The oxime was reduced and the enol triflate generated using Comin’s reagent. Carboxymethylation followed by lactonisation and epoxidation provided compound 5, ready for the end game.
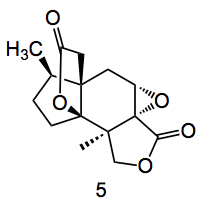
The two key reactions of the final sequence are

Reagents: TMSCl, LiN(TMS)2, -78°C then N-iodosuccinamide and finally 6 equivalents of DMDO in acetone at rt.
The formation of the γ-keto-lactone is interesting and apparently involves “an ostensible iodoso- Pummerer rearrangement“. This reaction had been previously described by Danishefsky and others. Danishefsky, himself, suggests that a number of mechanisms can be devised for this transformation:”including one which proceeds by an initial syn-elimination, followed by epoxidation of the highly strained double bond with subsequent rearrangement Alternatively, a more simplistic explanation would include tautomerization of the proposed iodoso intermediate (to form the iodo-analogue of an oxime), followed by hydrolysis“. Whatever the case the synthesis was completed by treatment of the epoxide with lithium hydroxide which gave the desired product in reasonable yield.
So there we have it a very nice approach to a complex natural product in 18 steps with a reasonable overall yield. The highlights for me were the use of the Robinson annelation, a reaction I haven’t done since university and the C-H oxidation method of Sanford which has apparently been applied for the first time in natural product synthesis. Congratulations on a nice synthesis and a very readable paper.
![]()